Finding Relief After Years of Unbearable Pain from Sickle Cell Disease: Allan's Story
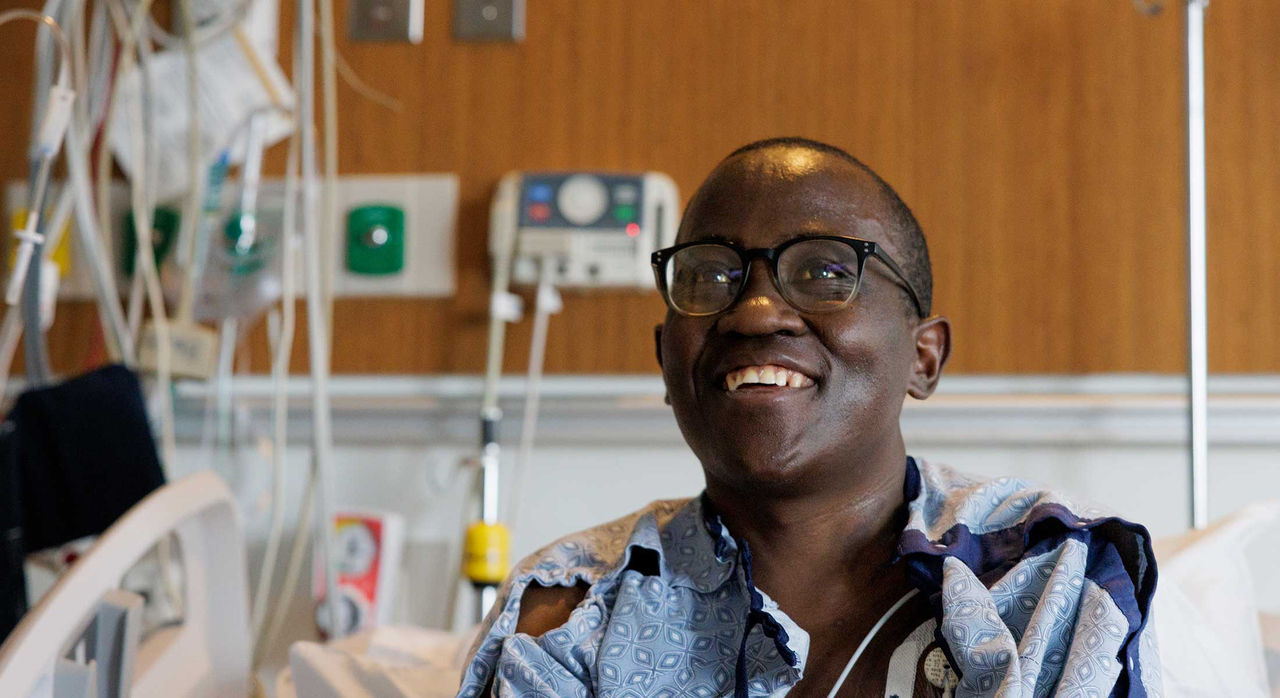
On a scale of 1 to 10, Allan Byamukama said, the pain was a 10. It could start anywhere and spread throughout the body mercilessly fast.
"Excruciating pain. It feels like your bones are being crushed. Within minutes, you can barely walk or talk," he said. "You always live in fear because you never know when the next pain crisis will come."
Allan, 34, is describing his experience living with sickle cell disease (SCD). For most of his life, he suffered through recurring pain crises caused by this blood disorder. Each one meant a trip to the emergency room (ER) to manage the pain.
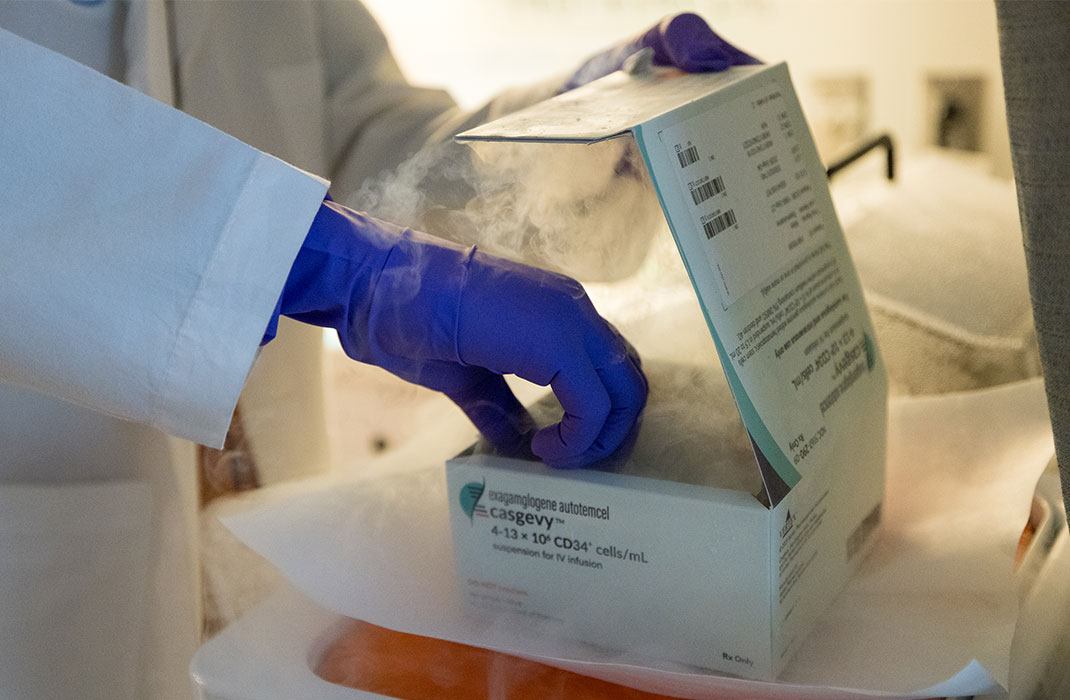
Two years ago, Allan became a patient at Massachusetts General Hospital’s Comprehensive Sickle Cell Disease Treatment Center. Recently, he became one of the first 15 people in the world to receive a cutting-edge therapy for SCD. While it's not a cure, it has significantly eased his symptoms—and turned his life around.
Diagnosed with SCD as an infant
SCD, which runs in families, affects around 100,000 Americans. It is caused by abnormal hemoglobin, the protein in red blood cells that carries oxygen to different parts of the body. The abnormal hemoglobin makes the red blood cells turn hard and sticky. The cells assume a "C" shape, like a sickle.
As the sickled cells move through blood vessels and organs, they can get stuck and block blood flow. This leads to pain that can range from mild to severe. Possible complications include stroke, heart attack, and potentially life-threatening organ damage. In addition, someone living with SCD has a lifespan two to three decades shorter than the average person.
Allan, who was born in Uganda, started showing signs of illness at five months. His eyes were yellow, his fingers and toes were often swollen, and he cried a lot. Tests at a local hospital found he had SCD.
Growing up, Allan knew he wasn't like the other kids. His mother didn't let him play soccer, fearing excessive activity would spark pain crises. Due to hospital stays, he missed long stretches of school.
The extreme pain arrived sporadically—sometimes every other month, other times four or five months apart. Whenever a crisis struck, his parents took him to the hospital to receive strong pain medications. "I'm thankful to them for their support and care," Allan said.
At age 28, Allan moved from Uganda to Worcester, Massachusetts. Beside continuing his education, he wanted to see if the region's renowned hospitals could tame his SCD.
Barriers to leading a normal life
During his first several years in Massachusetts, Allan saw a hematologist (blood doctor) at a local hospital. While working for a nonprofit organization, he also began pursuing a graduate degree in social work. But over time, his pain crises grew more frequent and intense. Eventually, he had to go to the ER every few days.
Then he decided to seek care at Mass General's sickle cell clinic. At one appointment, a doctor there mentioned the U.S. Food and Drug Administration (FDA) was about to approve a new gene therapy for SCD. Allan said if the opportunity ever arose, he would like to proceed with the treatment.
"I didn't even hesitate," he said. "I figured, if it could change my life, why not?"