CRISPR Gene Therapy for Sickle Cell Disease

According to the National Institutes of Health, more than 20 million people worldwide live with sickle cell disease (SCD), the most common inherited blood disorder in the United States. The condition affects Black patients far more than any other racial group, often leading to chronic pain and a shortened lifespan.
There is no readily accessible and widely available cure for the condition. However, recent developments in gene therapy, the practice of replacing or modifying a faulty gene to treat or stop disease progression, offer hope to patients living with the condition.
In December 2023, the U.S. Food and Drug Administration (FDA) approved the first two gene therapies for SCD: Casgevy and Lyfgenia. The Comprehensive Sickle Cell Disease Treatment Center at Massachusetts General Hospital is one of the first centers in the U.S. to administer Casgevy.
“When it comes to SCD, patients with the condition are too used to feeling like they’re being treated last,” says Sharl Azar, MD, a Mass General Brigham hematologist. “Thanks to the vast network of expertise we have across Mass General Brigham, we have the opportunity to help right this wrong.”
Dr. Azar serves as medical director of the Comprehensive Sickle Cell Disease Treatment Center. He explains how gene therapy works for SCD and how the treatment can combat racial inequities across health care.
What is gene therapy?
All species possess their own instruction manual for survival. These instructions, or genes, exist inside every living cell. Long strands of deoxyribonucleic acid, or DNA, are the figurative “sentences” inside the manual. Cells read, or transcribe, sections of DNA to produce proteins essential for daily life.
In humans, proteins can determine characteristics as harmless as eye color or as important as eyesight. A slight change in DNA, however, can drastically affect the ability to function. Changing a single gene can result in chronic, often debilitating conditions.
In recent years, doctors have developed gene therapy technology that can fix errors, or mutations, to DNA. One technology, called clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9, or CRISPR-Cas9, can add, remove, or alter specific locations of genes faster and more accurately than other gene-editing tools.
Researchers in the Mass General Brigham Gene and Cell Therapy Institute have explored several new applications for gene therapy technology, including for:
Deafness and blindness: Mass Eye and Ear researchers have developed CRISPR/cas9 models capable of targeting mutations responsible for deafness and blindness.
Cancer: Mass General Brigham researchers can harvest a patient’s immune cells and re-engineer them to recognize and kill cancer cells. This therapy, called CAR (chimeric antigen receptor) T-cell therapy, commonly treats advanced blood cancers, including certain leukemias, multiple myeloma, and non-Hodgkin’s lymphoma. Most recently, it has also showed promising results for treating glioblastoma.
Alzheimer’s disease: Brigham and Women’s Hospital researchers assessed whether manipulating a gene in the brain could help treat familial Alzheimer’s disease. Evidence collected from mice suggested the intervention may work one day.
Gene therapy for sickle cell disease
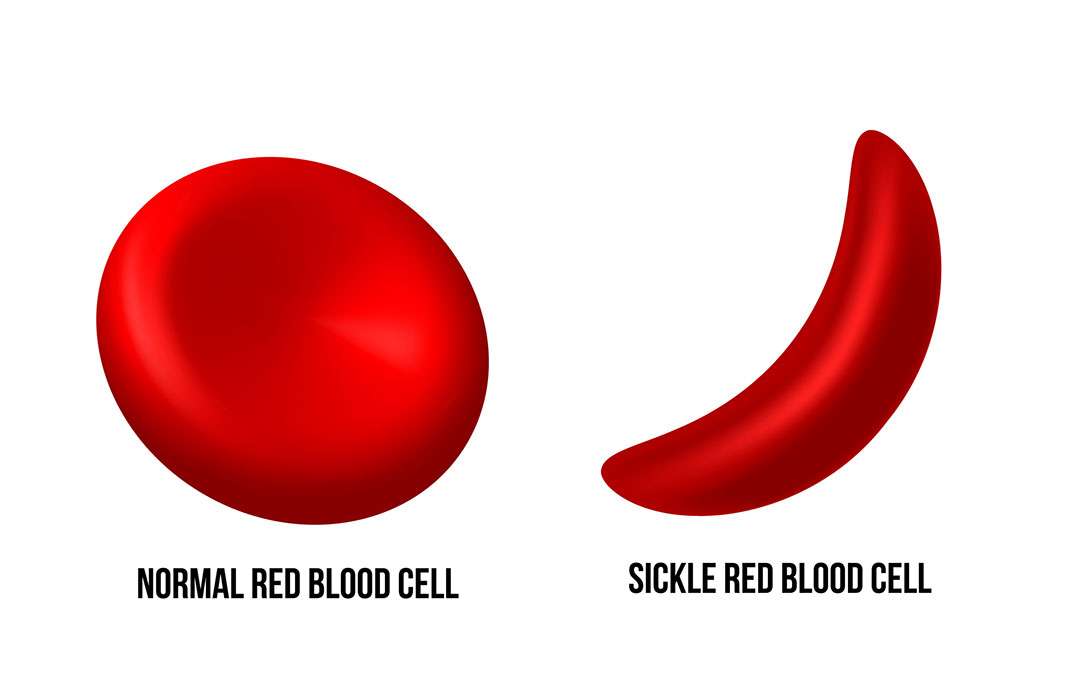
SCD occurs from a change, or mutation, to the figurative genetic sentence responsible for creating hemoglobin, a protein that allows red blood cells to carry oxygen throughout the body.
Healthy hemoglobin gives a red blood cell its unique shape: a circular, indented disc. The circular shape allows red blood cells to flow smoothly through blood vessels. The mutation responsible for SCD produces an abnormal protein, which changes the shape of red blood cells into a sickle, or crescent.
These “sickle” cells can easily break apart in the blood stream. According to the Centers for Disease Control and Prevention (CDC), the most severe cases of SCD can shorten the lives of patients by 20 to 30 years.
“The sickle shape is really only a part of the story,” says Dr. Azar. “The disease is driven by a host of damaging effects that come from this single genetic mutation. Finding a way to mitigate the negative effects of the mutation would profoundly affect a patient’s quality of life, let alone their lifespan.”
The two treatments approved by the FDA in December 2023 — Casgevy and Lyfgenia — use different approaches to gene therapy to treat red blood cells at the source. Each therapy modifies stem cells extracted from bone marrow, a spongy tissue found inside bones. These stem cells, which can transform into different cell types, ultimately grow into red blood cells.
Casgevy
Before a baby is born, bone marrow produces red blood cells with a kind of hemoglobin different from the kind found in children and adults. This hemoglobin, called fetal hemoglobin, serves the same function as regular hemoglobin, or hemoglobin A. After birth, bone marrow begins replacing red blood cells carrying fetal hemoglobin with those carrying hemoglobin A.
Different genes are responsible for producing each type of hemoglobin. The mutation for SCD only affects the production of hemoglobin A and not fetal hemoglobin.
Casgevy, produced by Vertex Pharmaceuticals with support from CRISPR Therapeutics, uses CRISPR/cas9 to restart the production of fetal hemoglobin. It is the first FDA-approved therapy using CRISPR/Cas9 technology.
Lyfgenia
Lyfgenia, produced by Bluebird Bio, Inc., uses neither CRISPR nor fetal hemoglobin to treat SCD. Instead, it uses a harmless virus called a lentiviral vector to deliver genetic material to stem cells. The genetic material modifies these cells to produce a hemoglobin that functions similarly to hemoglobin A called hemoglobin AT87Q.
“One method restarts a function that humans already had, while the other modifies a gene to produce a type of hemoglobin they couldn’t produce on their own,” says Dr. Azar.
How can gene therapy treat sickle cell disease symptoms?
Patients with SCD experience chronic pain throughout their lifetime.
Sickle cell hemoglobin makes a red blood cell harder, stickier, and less flexible. When trying to squeeze through narrow blood vessels, these sickle cells may stick together, or to the sides of vessels. The most severe pain, called pain crises, occur from sickle cells sticking to vessels and the ensuing damage they cause.
Other complications include:
Damage to certain organs, such as the heart, lungs, and kidneys
Casgevy and Lyfgenia make it less likely for red blood cells to obstruct blood flow or form a sickle shape. The fetal hemoglobin and hemoglobin AT87Q produced by the respective drugs restore much of the natural properties of these red blood cells.
How do doctors perform gene cell therapy for patients with sickle cell disease?
Racial inequity in sickle cell disease treatments
According to the American Red Cross, 98% of patients with SCD are Black or Brown. The few treatments available underscore racial inequity across health care, especially when compared to cystic fibrosis, another terrible genetic disease that mostly impacts white patients.
SCD is 3 times more common than cystic fibrosis. But, from 2008 to 2018, research shows that both diseases received a similar amount of federal government research funding. Journals published significantly more research articles about cystic fibrosis, and the FDA approved three more drugs for cystic fibrosis.
In 1998, the FDA approved the drug hydroxyurea to treat adults with SCD. According to the American Society of Hematology, the drug helps raise fetal hemoglobin levels and helps red blood cells stay round and flexible. Since then, progress had stalled.
“For 25 years, hydroxyurea was the only FDA-approved drug that could significantly help patients with sickle cell disease live longer and with less pain,” says Dr. Azar. “When the other two options are hardly available for a group disproportionately affected by the disease, there becomes an urgent need to find new pathways for treatment.”
The two alternative treatments are:
1. Blood transfusion
Patients with SCD may require one or more blood transfusions in their lifetime. A blood transfusion provides healthy blood from a donor to a recipient who:
May have lost blood through a surgery or injury
Can’t produce enough healthy blood on their own
Blood transfusions may take 1 to 4 hours to complete. Doctors must verify whether a recipient’s blood type matches that of a donor. Blood also must carry the same type of human leukocyte antigen (HLA), a protein found in bodily tissue.
The immune system, the body’s main line of defense from harmful diseases, uses HLA to flag normal, healthy tissues from harmful substances. Identifying the correct HLA prevents the immune system from attacking those healthy tissues. If blood type or HLA do not match, the recipient’s body may reject, or attack, the blood it receives.
Frequent blood transfusions can also lead to an overload of iron, which can make it more difficult to find matching blood in the future.
“Since blood transfusion is the only approach to treating sickle cell emergencies, it is imperative to reduce the barriers to finding a blood match,” says Dr. Azar.
2. Bone marrow transplant
During a bone marrow transplant, doctors replace abnormal stem cells with healthy stem cells from a donor. Patients stay in the hospital for 1 to 2 weeks before the procedure.
Special medications — and sometimes radiation — destroy the abnormal stem cells leftover in the patient’s bone marrow. They also weaken the immune system, preventing the patient’s body from attacking bone marrow from the donor. Like a blood transfusion, doctors must ensure the patient’s HLA and blood type match those of the donor.
The medications and radiation may lead to:
Nausea and vomiting
Diarrhea
Tiredness
Mouth sores
Skin rashes
Hair loss
Liver damage
People inherit HLA types, which means ethnicity can make it much more difficult to find a match for a blood transfusion or a bone marrow transplant. According to the Be The Match Registry®, white patients have a 79% chance of finding a bone marrow match. Black patients only have a 29% chance.
What centers offer gene therapy for sickle cell disease?
When successful, Casgevy and Lyfgenia allow bone marrow to create enough healthy red blood cells to limit the effects of sickle cells. According to the FDA, patients who underwent both treatments experienced significant relief from sickle cell vasoocclusive events (VOEs), or pain crises, in separate clinical trials.
Few medical centers nationwide offer either therapy. Vertex has authorized 9 centers to administer its treatment. Bluebird Bio has authorized 27.
Mass General meets several requirements for offering Casgevy. The requirements, set by the National Alliance of Sickle Cell Centers, include having:
A comprehensive SCD center
A bone marrow transplant center accredited by the Foundation for the Accreditation of Cellular Therapy
An apheresis unit, which helps separate the different components of blood
Social work, mental health care, nursing care, and nutritional support services
Expanding access to gene therapy for sickle cell disease
Dr. Azar says the Comprehensive Sickle Cell Disease Treatment Center at Mass General aims to make Casgevy more accessible to the thousands of patients who need it.
The center actively addresses accommodations for patients traveling long distances. Its clinicians have also advocated for bills in the Massachusetts State Legislature that would require insurance providers to cover the costs of fertility treatments.
Additional resources offered by the center include:
Individualized techniques for treating pain, such as yoga, massage therapy, and acupuncture
An SCD chaplain for spiritual care
A medical exercise specialist who can help to set personalized exercise and nutritional plans
Dedicated palliative care providers who can help strategize around symptom management and survivorship
Dedicated primary care providers who can provide preventative care and improve overall wellness
“The sad reality is that many sickle cell patients have been told they’re never going to live past their 40s, and never invested in a home or a retirement plan,” says Dr. Azar. “The power of gene cell therapy potentially gives us an opportunity to rewrite this narrative.”
